
Título
Autor
Fecha
Lugar de Realización
Texto
Revista Argentina de Neurocirugía, 15: 29, 2001
Actualización
Ecografía en el Diagnóstico de las Malformaciones Raquimedulares
José Ochoa y René Conci
Departamento de Diagnóstico por Imágenes Hospital Universitario de Maternidad y Neonatología, Universidad Nacional de Córdoba, Córdoba.
Correspondencia: Santa Rosa 748 (5000) Córdoba, Argentina
E-mail: josehochoa@infovia.com.ar
RESUMEN
La ecografia constituye actualmente el mejor método para la valoración fetal y el diagnóstico presuntivo de anomalías vertebrales y medulares. Realizado por personal entrenado e interpretado adecuadamente este estudio puede informar, en un estadio prenatal, la existencia de los distintos tipos de anomalía. Este conocimiento resultará de utilidad para la planificación de una terapéutica adecuada.
Palabras clave: disrafias, defectos del tubo neural, ecografia prenatal, malformaciones raquimedulares.
ABSTRACT
Ecography is the best diagnostic method for the study of prenatal spinal cord and vertebral fetal malformations. Performed by trained personal it can inform accurately about prenatal malformaciones. The knowledge of the images may be a useful guide to perform an appropriate treatment.
Key words: disraphy, neural tube defects, prenatal ecography, spinal malformaciones
EMBRIOLOGÍA
El cerebro, la médula espinal y el conducto raquídeo se desarrollan a partir de la placa neural en un proceso denominado "neurulación" y que comprende la formación y cierre del tubo neural embrionario.
Al final de la segunda semana de desarrollo (día 15-16 postconcepcional) el disco germinativo bilaminar (constituido solo por endodermo y ectodermo) comienza un proceso a través del cual se desarrolla la capa germinativa mesodérmica por invaginación de las células del ectodermo a nivel de la línea denominada primitiva14 (Fig 1).
En el extremo anterior de esta línea conocida como nudo de Hensen las células se invaginan en sentido cefálico a manera de tubo formando la notocorda o prolongación notocordal primitiva, la cual en un primer momento se une con el endodermo formando el conducto neurentérico, para luego separarse y constituir la notocorda definitiva. Este cordón macizo, probablemente a través de un proceso de inducción promueve cambios en el disco embrionario, ahora trilaminar, dando lugar a la formación del tubo neural. El proceso completo de formación y cierre ocurre aproximadamente en 12 días (Fig 2).
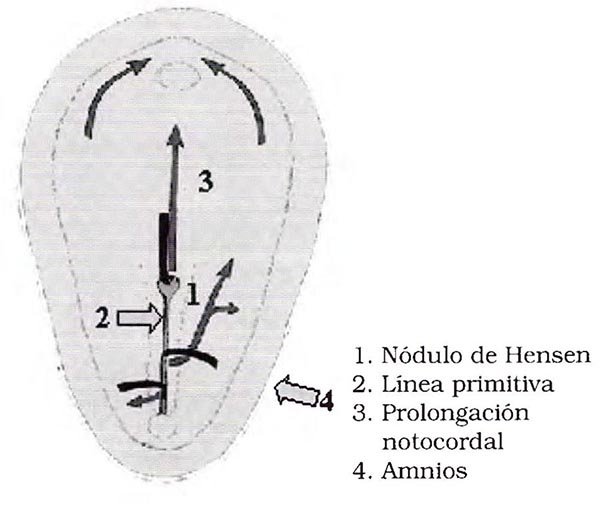
Fig 1. Día 16 postconcepcional (adaptado de Langman)14

Fig. 2. Día 18 a 26 postconcepcional (adaptado de Langman).14
Inicialmente la placa neural se invagina formando un surco (día 18 postconcepcional), para luego dar lugar a un tubo que se despega del ectodermo superficial cerrándose en sentido cefálico y caudal. El tubo queda inicialmente en comunicación con la cavidad amniótica por sus extremos a través del neuroporo anterior (cefálico) que se cierra el día 24 o 25 postconcepcional y del neuroporo posterior (caudal) que lo hace entre los días 26 a 28, cuando el embrión tiene tan sólo 5 mm de longitud cráneo caudal14,19,20.
Una falla de este proceso del desarrollo da lugar a una de las anomalías fetales más frecuentes, los defectos de cierre del tubo neural (DCTN).
INCIDENCIA
La espina bífida es la anomalía más común del sistema nervioso central con una incidencia entre 0,3 y 3,5 por mil nacidos vivos9,19. Varía según el área geográfica, existen diferencias étnicas e incluso se han reportado variaciones estacionales. El riesgo de defecto de cierre del tubo neural se incrementa a 20-30 por mil nacimientos si previamente existía otro hijo afectado20.
CLASIFICACIÓN
Disrafismo espinal es el término aplicado para describir una variedad de anomalías de la columna vertebral cuyo común denominador es el cierre incompleto de las tejidos neurales, óseos o mesenquimáticos de la línea media.
El término espina bífida se refiere al cierre incompleto de los elementos posteriores de la columna vertebral (lámina y procesos espinosos). La clasificación inicial de Harwood-Nash, Naidich y McLone, luego modificada por Byrd et al3, señala 3 categorías (Tabla 1).
Tabla 1. Disrafias espinales

En la espina bífida abierta (aperta) la placa neural se encuentra expuesta y unida a la piel por sus márgenes o por medio de la píaaracnoides, que la recubre internamente. La placoda puede encontrarse al mismo nivel que la piel (mielocele) o sobresalir posteriormente (mielomeningocele).
El segundo grupo de anomalías incluye aquéllas cubiertas por piel pero que presentan un abultamiento posterior, encontrándose en esta categoría el lipomielomeningocele, el mielocistocele y el meningocele posterior.
La espina bífida oculta comprende un número mayor de anomalías (Tabla 2). Si bien no existe masa posterior a veces pueden observarse algunos signos cutáneos sugestivos como un "mechón de pelo", hoyuelo, nevus, hiperpigmentación o angioma. Otros signos clínicos como el pie bot, disturbios en la marcha, paresias, disfunción esfmteriana, etc. pueden alertar también sobre esta posibilidad.
Tabla 2. Espina bífida oculta

ETIOPATOGENIA
La mayoría de los DCTN aparecen como una condición aislada probablemente multifactorial en individuos genéticamente normales19. Pueden también ocurrir como parte de aneuploidías o síndromes Mendelianos (Tabla 3).
El riesgo de recurrencia se estima de 5% para futuros embarazos y si existen dos hermanos afectados alcanza un 13%.
Tabla 3. Defectos de cierre del tubo neural

PREVENCIÓN
El uso periconcepcional de ácido fólico reduce considerablemente el riesgo de DCTN. Se ha recomendado para las madres en edad de procrear la ingesta de 0,4 mg (400 mcg) diarios de ácido fólico. Aquéllas que ya tuvieron un hijo afectado deben aumentar la dosis a 4 mg /día comenzando por lo menos un mes antes de la concepción5.
DIAGNÓSTICO
El diagnóstico prenatal depende de la demostración del defecto espinal a través de la ecografía. La ecografía intravaginal a las 13-14 semanas o transabdominal a partir de la semana 15-16 de gestación permiten el diagnóstico del defecto espinal.
En la década del 80 el modo principal de descarte de los DCTN fue la alfafetoproteína sérica materna alrededor de la semana 16 de gestación y la determinación de alfafetoproteína y acetilcolinesterasa en líquido amniótico25. Este diagnóstico no era sin embargo específico ya que se observó que un gran número de alteraciones fetomaternas puede también elevar los niveles de alfafetoproteína (Tabla 4).
Tabla 4. Causas de elevación de AFP

El perfeccionamiento de la resolución de los equipos de ultrasonografía tiende a reemplazar el uso de la alfafetoproteína, tanto como método de screening como para confirmación diagnóstica, demostrando gran confiabilidad25. Su realización requiere, sin embargo, operadores entrenados y exámenes dirigidos, para alcanzar su máximo potencial17. En un estudio multicéntrico la ecografia de rutina realizada entre las 16 y 22 semanas de gestación alcanzó una sensibilidad superior al 90% para el diagnóstico de espina bífida22.
El diagnóstico ultrasonográfico se basa en la demostración del defecto espinal y en signos secundarios a nivel craneal que frecuentemente acompañan a la espina bífida.
La visualización de las partes blandas resulta dificil en fetos pequeños ya que son muy delgadas, por lo que excepto en casos de masas espinales de un volumen significativo, el diagnóstico se apoya en gran medida en la demostración de los núcleos de osificación espinales, que son tres a saber: uno anterior que corresponde al centro del cuerpo vertebral (ver hemivértebra) y dos procesos laterales a nivel de los pedículos y de las láminas, en la base de las apófisis transversas. La osificación comienza en la semana 10 de gestación y va progresando en sentido cefalocaudal hasta alcanzar las últimas vértebras lumbares en la semana 13 aproximadamente. Se ha informado evaluación satisfactoria de los centros de osificación mediante ecografía combinada transabdominal e intravaginal y también de otros signos craneales que pueden permitir un diagnóstico precoz de espina bífida en el primer trimestre25.
Al avanzar la gestación los núcleos de osificación se van haciendo cada vez más prominentes, mientras que las partes blandas tienen mayor espesor, y si bien existen variaciones individuales entre feto y feto, en general se puede aseverar o descartar el diagnóstico de espina bífida en las semanas 18 a 20 de gestación utilizando equipos de adecuada resolución. Se pueden utilizar innumerables planos de corte para estudiar la columna vertebral. Los planos clásicos permiten una adecuada demostración de los elementos anatómicos citados.
En el plano axial se observan los tres núcleos de osificación, se puede medir el canal espinal y la distancia entre las láminas y pedículos posteriores; el plano sagital muestra la columna en sentido longitudinal y el plano coronal valora la separación de los pedículos, el diámetro del canal espinal. Todos otorgan, cuando existe una adecuada cantidad de líquido amniótico buena visualización de las partes blandas suprayacentes (Figs. 3 y 4).
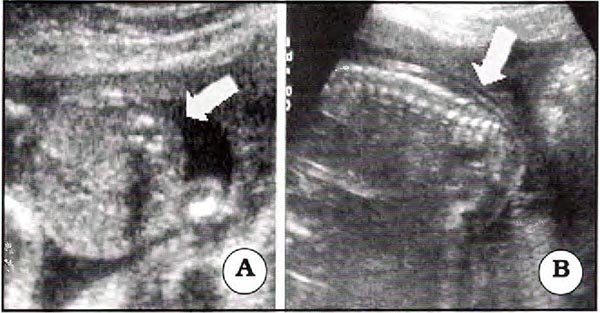
Fig. 3. Planos de corte. A. Axial. B. sagital

Fig. 4. Plano de corte coronal
ESPINA BÍFIDA ABIERTA
La forma abierta constituye la mayoría de los casos de espina bífida. Los pliegues del canal neural no se fusionan en la línea media para constituir el tubo neural y permanecen adheridos con el ectodermo cutáneo y por lo tanto la placa neural permanece unida a la piel y no permite la migración del mesodermo hacia la línea media. Por detrás de la placoda neural existe tejido meníngeo conteniendo líquido cefalorraquídeo (pía y aracnoides). Se denomina mielocele cuando la placoda neural se encuentra expuesta y no protruye. Cuando el espacio subaracnoideo se encuentra ensanchado y desplaza posteriormente la placoda, la cual abomba, constituye mielomeningocele2. El diagnóstico ecográfico se basa en la demostración de los signos consignados (Figs. 5 a 11)

Fig. 5. Las flechas muestran el defecto espinal sín protrusión (mielocele) entre L2 y S3. Los números indican vértebras lumbares.

Fig. 6. Vista axial de un cuerpo vertebral a nivel de la raquisquisis lumbar. Se evidencia defecto constituido por delgada membrana que no protruye. Mielocele

Fig. 7. Vista axial: apertura y separación de los pedículos vertebrales. Las flechas señalan la delgada lámina de tejido neural. Mielocele

Fig. 8. Vista longitudinal de la columna vertebral demostrando mielocele.

Fig. 9. Vistas coronales. Raquisquisis lumbosacra en 3 pacientes diferentes, demostrando el ensanchamiento del canal espinal y la separación de los pedículos posteriores.
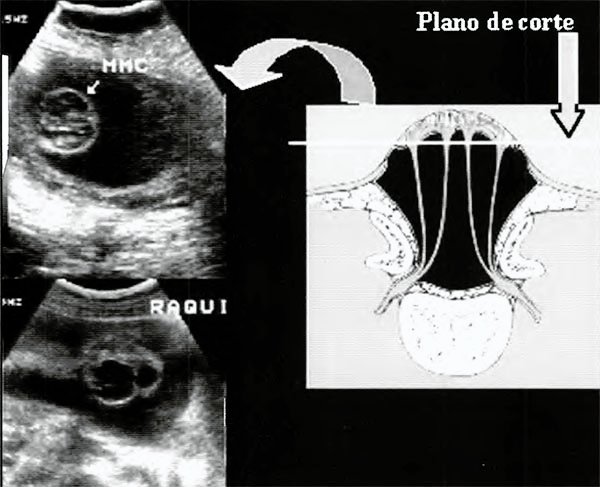
Fig. 10. Vistas coronales a nivel del defecto. Mielomeningocele: protrusión del tejido neural.

Fig. 11. Tres vistas diferentes del mielomeningocele. Se visualiza la placoda neural unida a la piel por meninges. Los tabiques representan raíces nerviosas que nacen en la placa neural.
ANOMALÍAS CRANEALES ASOCIADAS A ESPINA BÍFIDA
En los casos de espina bífida abierta la médula espinal se encuentra anclada dando lugar, en un porcentaje superior al 95%, a la anomalía de Chiari tipo II que se caracteriza por una herniación del vermis cerebeloso a través del foramen magno, juntamente con el cuarto ventrículo. Hay además descenso de la tienda del cerebelo y disminución del tamaño de la fosa posterior. También existe dismorfia ventricular que afecta los ventrículos laterales y tercer ventrículo con receso pineal amplio y masa intertalámica prominente. En la gran mayoría de los casos la anomalía de Chiari se acompaña de hidrocefalia obstructiva2,3,13,19,20. Estas alteraciones craneales pueden ser detectadas tempranamente en la gestación22,25 y son orientadoras hacia la presencia de un defecto espinal. Rutinariamente se investiga a nivel cerebral la línea media y la presencia de quistes del septum (cavum septum pellucidum), los ventrículos cerebrales y la cisterna magna. Además se analiza la conformación de la calota craneal. El examen adecuado de estas estructuras permite un elevado porcentaje de sospecha diagnóstica. La obliteración de la cisterna magna y la compresión del cerebelo que adopta una forma semicircular (denominada gráficamente "signo de la banana"), la dilatación de los ventrículos laterales y la deformidad de la calota craneal a nivel frontal ("signo del limón") son alteraciones estructurales orientadoras a la presencia de defecto espinal, aún cuando el feto sea pequeño y/o la visualización de la columna vertebral resulte dificultosa.
Las figuras 12 a 15 muestran las alteraciones descriptas.
Entre las anomalías extraespinales y extracraneales asociadas, las más frecuentes son las correspondientes a los miembros inferiores así como las alteraciones renales secundarias o no a la presencia de vejiga neurogénica (Tabla 5).
Tabla 5. Anomalías extracerebrales asociadas

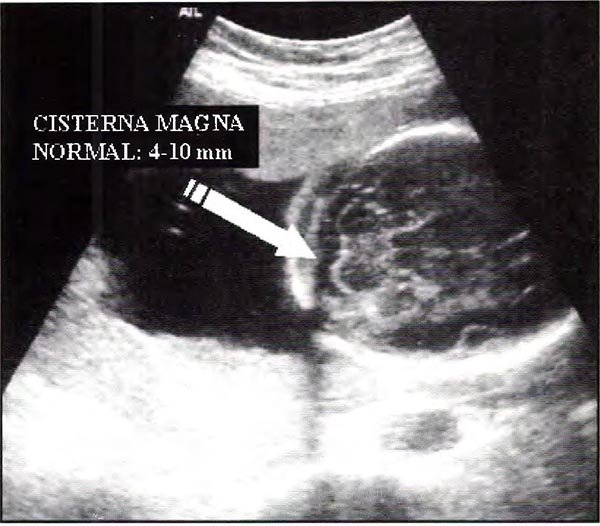
Fig. 12. Vista axial posterior del cráneo demostrando la visualización normal de la cisterna magna y sus valores normales.
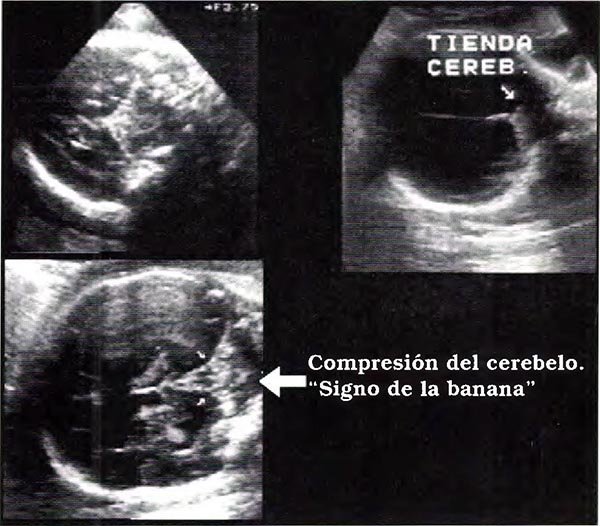
Fig. 13. Vistas axiales y coronales posteriores en pacientes con mielomeningocele. Se observa descenso de la tienda y del cerebelo con obliteracion de la cisterna magna. El cerebelo adopta una forma semicircular "signo de la banana".
La espina bífida abierta suele asociarse a pie equino varo19,20 con trastornos tróficos representados por una menor masa muscular producto de la afectación de los nervios periféricos (Fig. 16 A). Por la misma razón y el disbalance muscular existente estos niños pueden presentar displasia del desarrollo de la cadera en período pre o postnatal.

Fig. 14. Vistas axiales. Existe dilatación de diverso grado, a veces con crecimiento ventricular asimétrico. La determinación del índice ventrículo cortical permite el seguimiento prenatal.

Fig. 15. Deformidad frontal que constituye el denominado "signo del limón" en vistas sagital y axial.
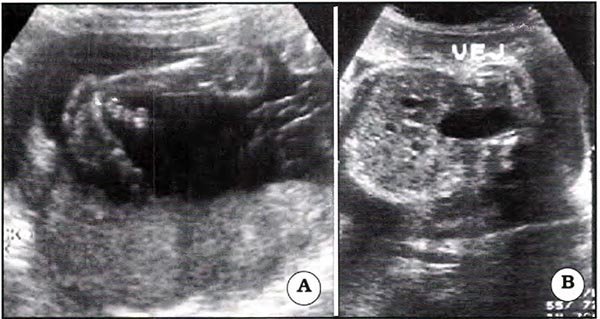
Fig. 16. A Pie bot. El plano de visualización del pie corresponde al eje de la pierna. B. Vejiga elongada, con disminución del diámetro transversal, persistentemente llena.
La vejiga urinaria está casi invariablemente afectada, (Fig. 16B), y se encuentra persistentemente llena, en ocasiones con disminución del diámetro transversal y forma elongada. Secundariamente puede dilatarse la vía urinaria superior en presencia de reflujo u obstrucción relativa. Hemos observado además displasia renal acompañando a mielomeningocele (Fig. 17).
Las tablas 6 y 7 y las figuras 18 y 19 muestran las alteraciones asociadas a espina bífida abierta.
Tabla 6. Anomalías espinales asociadas

Tabla 7. Otras anomalías fetales

ANOMALÍA DE CHIARI TIPO III
Es una rara condición caracterizada por herniación del contenido de la fosa posterior y espina bífida cervical alta ubicada a nivel de C 1 - C22.
La anomalía suele acompañarse de cefalocele occípitocervical a través del cual puede herniarse el cerebelo displásico, 4° ventrículo y en ocasiones tallo encefálico.
El diagnóstico prenatal puede ser realizado al detectar raquisquisis cervical alta y descenso de los elementos de la fosa posterior. La figura 20 muestra las imágenes que se obtuvieron en el segundo trimestre en un feto afectado por esta infrecuente anomalía.

Fig. 17. A. Riñón con incremento de la ecogenicidad y cambios quísticos asociado a mielomeningocele. B. Moderada ectasia renal asociada a vejiga neurogénica. Esta condición puede ser también un hallazgo en fetos normales o con uropatía no neurogénica

Fig. 18 A. Las flechas juntas señalan cifoescoliosis marcada. B Las flechas separadas indican protrusión por mielomeningocele. PLAC: placenta

Fig. 19. Vista sagital de columna cérvico dorsal. Pequeño quiste intraespinal que comprime la médula en unfeto de 28 semanas con mielomeningocele lumbosacro.
ESPINA BÍFIDA OCULTA
El diagnóstico prenatal de la espina bífida oculta es sin duda más dificil que en su variedad abierta y en ocasiones pueden pasar inadvertidas en el examen fetal. La ecografía postnatal puede en ocasiones ser de gran ayuda en la valoración de este tipo de defectos. La tabla 8 muestra un grupo heterogéneo de anomalías que se incluyen dentro de esta denominación.
DIASTEMATOMIELIA
Implica una fisura sagital que divide el cordón espinal en dos hemimédulas. Cada una de ellas muestra un canal central aunque sólo un asta dorsal y un asta ventral2. Puede ocurrir que el saco dural y la aracnoides estén también divididos en dos tubos que suelen estar separados por tabique óseo o cartilaginoso, o bien que exista un solo túnel dural y un solo espacio subaracnoideo que contenga las hemimédulas que están divididas por un tabique fibroso2. Puede ocurrir que comprometa a un único segmento vertebral o que abarque varias vértebras. La hendidura puede encontrarse a cualquier nivel, pero en la mayoría de los casos se encuentra a nivel de las ultimas vértebras dorsales o primeras lumbares.
La tabla 9 muestra los defectos comúnmente asociados a diastematomielia7. Entre los nevus cutáneos el más característico es el piloso, con largos cabellos situados a nivel del defecto espinal. El pie bot está asociado en la mitad de los casos. Este defecto espinal puede también asociarse a hidromielia16. Cuando acompaña a mielomeningocele, la hendidura sagital puede ocurrir al mismo nivel, superior o inferior a la raquisquisis y comprometer a una (denominada hemimielocele) o ambas hemimédulas. Hay predominancia en niñas (3:1).
Tabla 8. Espina bífida oculta

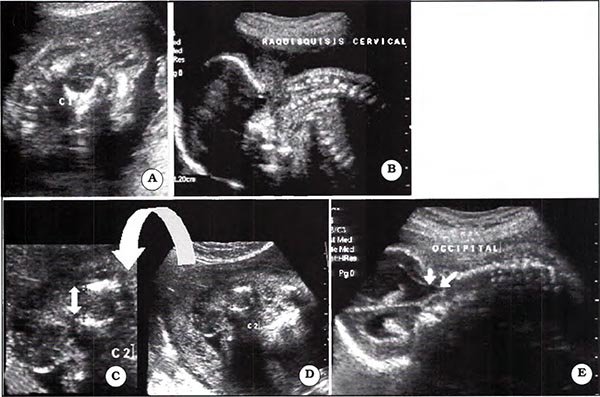
Fig. 20 A. Vista axial posterior. Se evidencia separación de pedículos a nivel de CI . B. Vista sagital demostrando el defecto espinal y descenso de los elementos anatómicos de fosa posterior. C. Vista ampliada de D muestra cerebelo dentro del defecto espinal, la flecha de doble cabeza señala la separación de pedículos en C2. E. Vista oblicua posterior muestra hidrocefalia y descenso del asta occipital del ventrículo lateral que se proyecta en el foramen magno.
Tabla 9. Diastematomielia: asociaciones

La columna vertebral y la médula espinal tienen similar longitud hasta el tercer mes de vida intrauterina. A partir de ese momento la velocidad de crecimiento de la columna vertebral supera a la de la médula, lo cual desplaza en sentido cefálico el cono medular. Éste se encuentra al nacer a nivel del extremo superior de L2 y a los 5 años a nivel del margen inferior de la misma vértebra. Si la médula se encuentra anclada la tracción de los elementos puede significar déficit neurológico progresivo de los miembros inferiores, que podrían beneficiarse de un diagnóstico temprano.
El diagnóstico prenatal del defecto oculto puede ser dificultoso, aún con operadores experimentados, si no existen alteraciones óseas asociadas. Se basa en el ensanchamiento del canal medular en vista coronal, tejidos blandos suprayacentes normales y en la demostración del tabique por lo que será más probable cuando exista tabique óseo o cartilaginoso. La estructura se observa como un foco ecogénico adicional ubicado entre las láminas posteriores. Éste es un hallazgo altamente específico y ha sido confirmado en todos los casos descriptos, luego del nacimiento1,23,28 (Figs. 21 a 24).
SENO DÉRMICO DORSAL
Deriva de una separación incompleta del ectodermo cutáneo y neural. Un delgado tubo tapizado por células epiteliales se invagina desde la piel alcanzando los tejidos subcutáneos. En el 50-66% de los casos tienen extensión intraespinal alcanzando la duramadre, la aracnoides o atravesándolas hasta alcanzar una raíz nerviosa, filum terminal o cono medular21. Se asocian frecuentemente con quistes dermoides o epidermoides que representan inclusiones embrionarias de la piel en el momento del cierre del tubo neural26. Pueden o no acompañarse de anomalías óseas como apófisis espinosa bífida, espina bífida oculta o defecto de las láminas8.
La ecografia postnatal puede demostrar el trayecto hipoecogénico que se desprende de la piel profundizándose hacia el canal espinal y puede servir como estudio inicial, utilizando transductores de alta resolución (Figura 25).
Tanto el lipoma intradural como los quistes dermoides y epidermoides del canal espinal son de dificil diagnóstico prenatal cuando no se asocian con alteraciones vertebrales al no realizarse en las ecografías de rutina (nivel 1) el examen del contenido espinal. La hidromielia es de visualización dificultosa a través de éste método de diagnóstico, siendo la resonancia magnética el mejor método de visualización.

Fig. 21. A. Vista sagítal muestra el tabique óseo de ubicación medial. B Vista axial señalando el tabique óseo y el ensanchamiento del canal espinal C. Vista coronal posterior demostrando tabique ecogénico en el canal espinal. AO: aorta materna.

Fig. 22. Estudio ecográfico postnatal de otro paciente con diastematomielia. A. Vista axial que demuestra la separación de los pedículos posteriores (flechas) por tabique ecogénico. B. En un corte inmediatamente superior se observan las hemirnédulas rodeadas de líquido. C y D. Ambos cordones medulares contenidos en un saco. El área anecogénica que los rodea corresponde a líquido cefalorraquídeo.

Fig. 23. Gestación de 31 semanas. Diastematomielia asociada a mielomeningocele. A. Amplia separación de los pedículos posteriores y la presencia de espolón óseo (flecha). B. Vista sagital. demostrando el espolón óseo desplazado en sentido posterior y rodeado del líquido anecogénico contenido en el mielomeningocele. C. Se visualiza en corte coronal la separación de las láminas posteriores, los delgados septos correspondientes al mielomeningocele y el espolón óseo interpuesto.

Fig. 24. RM postnatal. Diastematomielia asociada a mielomeningocele. A. Cifoescoliosis y vértebra en mariposa (flecha). B. Hemimédulas de gran longitud. La flecha señala la altura del espolón. C y D. Diastematomielia, RM. Vistas axiales a nivel del tabique que separa las hemimédulas.

Fig. 25. Estudio ecográfico en lactante menor con seno dérmico dorsal . A. Sagital oblicua que muestra estrecho canal anecogénico, en su extremo proximal. B. La flecha mayor destaca su comunicación con el canal espinal. M: musculatura paraespinal. CE: Canal espinal. C. Espina bífida oculta asociada al seno dérmico dorsal. Cuerpo vertebral inmediatamente superior al defecto muestra laminas posteriores que se unen en la línea media con apófisis espinosa cartilaginosa (anecogénica). D. Láminas posteriores (PP) que se encuentran separadas a nivel de la espina bífida.
OTRAS ANOMALÍAS VERTEBRALES
La cifosis y escoliosis son susceptibles de ser diagnosticadas en periodo prenatal.
La escoliosis congénita se produce por una falla en la formación, una falla en la segmentación o una mezcla de ambas10. Entre las anomalías comunes que causan escoliosis congénita se cuentan hemivértebras, vértebras en mariposa, barras de fusión, bloques vertebrales y hemivertebrales30.
Puede asociarse como se ha expresado previamente a espina bífida o coexistir con frecuencia defectos intraespinales ocultos como médula anclada, siringomielia, lipoma y diastematomielia18. Son frecuentes las anomalías cardíacas, renales y gastrointestinales asociadas. También puede formar parte de síndromes como Jarcho Levin y Klippel Feil o de la asociación VACTER.
En la sexta semana de gestación se desarrollan dos centros laterales de condrificación en los cuerpos vertebrales, los cuales rápidamente se unen (semanas 7-8) dando lugar al centro primario de osificación del cuerpo vertebral. Cuando uno de estos centros de condrificación no se forma se desarrolla una hemivértebra15.
La hemivértebra se puede presentar aislada o múltiple con una frecuencia de 5-10:10 000 nacimientos y con una relación varón-mujer de 1-2 a 1-329. La incidencia de defectos cromosómicos parece ser baja, cuando se trata de una anomalía aislada31.
El estudio ultrasonográfico muestra inadecuada alineación de los cuerpos vertebrales y escoliosis de diverso grado. Puede ser dificil su distinción de vértebra en mariposa o barra vertebral, pero un análisis meticuloso en vista coronal puede permitir la diferenciación de un elemento óseo incompleto interpuesto (Figs. 26 y 27).
SÍNDROME DE REGRESIÓN CAUDAL
También denominado displasia caudal es un defecto congénito caracterizado por ausencia de sacro, defectos de la columna lumbar y anomalías asociadas de otros sistemas, especialmente genitourinario, gastrointestinal y nervioso30. Hay posición alta del cono terminal, estrechamiento del canal espinal y del saco dural.
Cuando se asocia con malformaciones urogenitales y anorrectales como ano imperforado, malformación cloacal o extrofia de cloaca hay alta incidencia de disrafismo espinal como meningocele sacro anterior, lipomielomeningocele, mielocistocele o médula anclada14,30.

Fig. 26. Gestación de 28 semanas. Feto con asociación VACTER. Las flechas señalan la presencia de hemivértebras toracolumbares múltiples, sin mayor desviación del eje vertebral.
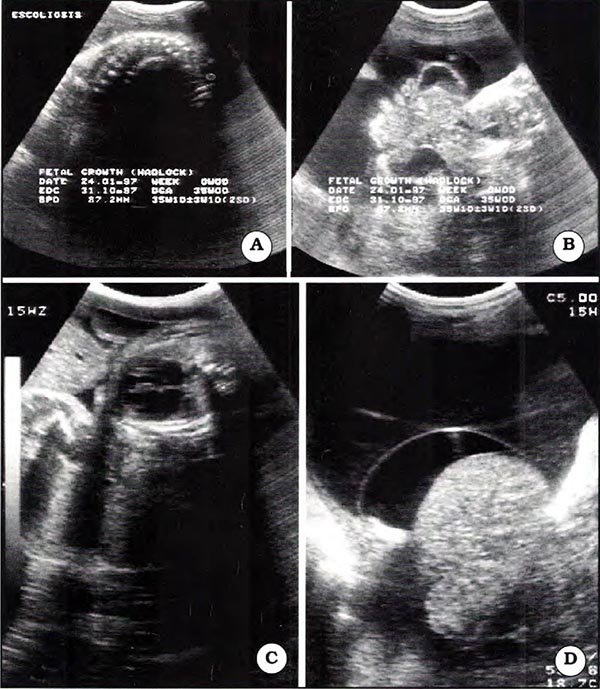
Fig. 27. Feto de 35 semanas. A. Marcada escoliosis vertebral. B. Defecto de la pared abdominal con protrusión de intestino cubierto por delgada membrana en la cavidad amniótica. C. Tórax fetal pequeño con alteración de la relación cardiotorácica. D. Hígado cubierto por membrana amnioperitoneal, por ausencia de la pared abdominal.
Su patogénesis no es clara pero se reconoce su asociación a diabetes materna11.
El diagnóstico prenatal va a depender de la magnitud del defecto y sus asociaciones. El aspecto más característico es la terminación brusca de la columna vertebral en vistas sagital y coronal, con aproximación de las alas ilíacas, con disminución del interespacio entre los fémures.
Secuencia Sirenomelia
Es considerada por algunos como el extremo más severo de la displasia caudal. Es una rara anomalía caracterizada por fusión de las extremidades inferiores que le dan al feto un aspecto sirenoide. Se cree que el defecto se origina de una falla en el desarrollo de las somitas caudales por teratógenos u otros factores. Entre ellos se ha mencionado la isquemia por "robo vascular11,27.
Se caracteriza por fusión completa de extremidades inferiores, imperforación anal, genitales externos ausentes o ambiguos, arteria umbilical única, agenesia renal, oligoamnios severo, defectos cardíacos, de pared abdominal y vertebrales11,24.
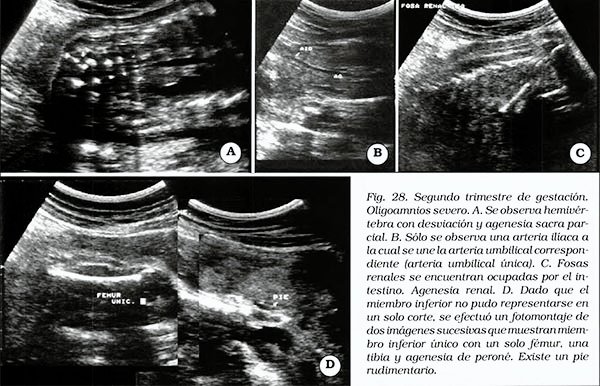
El diagnóstico ecográfico se sospecha ante la presencia de oligoamnios severo con agenesia renal. En estos casos se deberá examinar la columna vertebral en busca de anomalías lumbosacras y analizar los huesos largos de las extremidades inferiores. Ayuda también en la sospecha la presencia de arteria umbilical única24 (Figura 28).
CONCLUSIONES
La ecografia constituye en la actualidad el mejor método para la valoración fetal y el descarte de anomalías vertebrales y del SNC. Es preciso, sin embargo, que las ecografías obstétricas de rutina (nivel 1) sean realizadas por operadores entrenados en el rastreo de anomalías y que el examen fetal sea completo.
Ante la sospecha de anomalías debe practicarse un estudio dirigido o especializado, que permita además realizar los estudios fetales adecuados para llegar a un diagnóstico completo, comprendiendo todos los sistemas orgánicos (nivel 3).
La ecografía prenatal permite el diagnóstico de la gran mayoría de los defectos de la columna vertebral fetal. Para ello los estudios deben ser realizados en un periodo gestacional adecuado a fin de facilitar un examen en diferentes planos de visualización.
El uso de transductores lineales de alta resolución permite un examen adecuado en el neonato y lactante pequeño dado que aún no se ha completado la osificación de los elementos posteriores de la columna vertebral.
Si bien su utilidad no está aún completamente dilucidada la ecografia tridimensional 3D, podría representar un adelanto, al menos para una mejor demostración de los elementos defectuosos de la columna vertebral.
Bibliografía
1. Anderson NG, Jordan S, McFarlane MR, Lovell-Smith M: Diastematomyelia: diagnosis by prenatal sonography. Am J Roentgenol 163: 911-914,1994.
2. Barkovich A.J. Pediatric Neuroimaging 2a. Ed.z 1996; Lippincott Rayen Publishers, Philadelphia
3. Byrd S., Darling C., McLone.: Developmental Disorders of the Pediatric Spine, Radiol Clin North Am 29: 711-752, 1991.
4. Coerdt W. et al: Neural tube defects in chromasomically normal and abnormal human embryos. Ultrasound Obstet Gynecol 10: 410-415, 1997.
5. Committee on Genetics. American Academy of Pediatrics. Folie acid for the prevention of neural tube defects. Pediatrics 104: 325-327,1999.
6. den Hollander et al: Early tranvaginal ultrasonographic diagnosis of Beemer Langer dysplasia. A report of two cases. Ultrasound Obstet Gynecol 11: 298-302, 1998.
7. Guth Kelch AN, Jones RA, Zierski Diastema-tomyelia. Dev Med Child Neurol (Suppl) 13: 137138, 1971.
8. Harwood-Nash D. Fitz C. Neuroradiology in infants and children. 1976; CV Mosby, St Louis.
9. Holmes L. Driscoll S. and Atkins L.: Etiologic heterogeneity of neural tube defects. N Engl J Med 294: 365-369, 1976.
10. Jaskwhich D, Ali RM, Patel TC, Green DW Congenital scoliosis. J Pediatr Orthop 20: 61-66, 2000.
11. Jones KL. Caudal dysplasia sequence in Smith's recognizable patterns of human malformation. WB Saunders Company, 1998, Philadelphia.
12. Kennedy D et al. Inverted duplication of the distal short arm of chromosome 3 associated with lobar holoprosencephaly and lumbosacral meningomye-locele. Am J Med Genet 91: 167-170, 2000.
13. Kirpekar M., Cohen H.: Ultrasonography of the Neonatal Spine in Trimor-Tritsch's "Ultrasonography of the Prenatal and Neonatal Brain" la. ed.: Appleton & Lange, 1996, Conneticut.
14. Langman J. Embriología Médica. Desarrollo humano normal y anormal. 2a. ed. Nueva Editorial Interamericana, 1969, Mexico.
15. Moore KL, Persaud TVN: The Developing Human, 5a. ed. WB Saunders, Philadelphia 1993; pp 358364.
16. Pang D. Split cord malformation: clinical syndrome. Neurosurgery 31: 481-500, 1992.
17. Pilu G et al. Ultrasound of the fetal central nervous system. Curr Opin Obstet Gynecol 12: 93-103, 2000.
18. Prahinski JR et al.: Occult intraspinal anomalies in congenital scoliosis. J Pediatr Orthop 20: 59-61, 2000.
19. Romero Retal. Diagnosis of Congenital Anomalies Appleton & Lange Conneticut. 1988.
20. Sauberbrei E, Toi A. Columna fetal en Rumack C, Wilson S, Charboneau JW. Ecografía Obstétrica y Fetal.; Marbán Libros S.L., 2000, Madrid.
21. Schwartz H. Congenital tumors of the spinal cord in infants. Ann Surg 136: 183-192, 1952.
22. Sebire N.J. et al: Presence of the "lemon" sign in fetuses with spina bífida at the 10-14 week scan. Ultrasound Obstet Gynecol 10: 403-405, 1997.
23. Sepulveda W, Kyle PM, Hassan J, Weiner E: Prenatal diagnosis of diastematomyelia: case reports and review of the literature. Prenat Diagn 17: 161-165, 1997.
24. Sirtori M. Ghidini A, Romero R, HobbinsJC: Prenatal diagnosis of sirenomelia. J Ultrasound Med 8: 83-88, 1989.
25. Souka A.P. and Nicolaides K.H. Diagnosis of fetal abnormalities at the 10-14 week scan. Ultrasound Obstet Gynecol 10: 429-442, 1997.
26. Sternberg et al. Diagnostic Surgical Pathology. 3a. ed.; Lippincott Willams and Wilkins, 1999, Phila-delphia.
27. Stevenson RE, Jones KL, Phelan MC et al: Vascular steal: the pathogenetic mechanism producing sire-nomelia and associated defects of the víscera and soft tissues. Pediatrics 78: 451-457, 1986.
28. Winter RK, McKnight L, Byrne RA, Wright CH: Diastematomyelia: prenatal ultrasonic appearances. Clin Radiol 40: 291-294, 1989.
29. Wynne-Davies, R: Congenital vertebral anomalies: aetiology and relationship to spina bifida cystica. J Med Genet 12: 280-288, 1975.
30. Young Poussaint T, Barnes PD, Ball WS: Spine and Spinal Cord. In Kirks DR, Griscom NT, Practical Pediatric Imaging: Diagnostic Radiology of Infants and Children. 3a. ed., Lippincott Rayen Publ. 1998, Philadelphia.
31. Zelop CM, Pretorius DH, Benacerraf BR: Fetal Hemivertebrae: Associated anomalies, significan-ce, and outcome. Obstet Gynecol 81: 412-416, 1993.